Nel 2008 si è verificata una rivoluzione nel mondo della scienza, forse passata inosservata ai più.
Il progetto di sequenziamento del genoma umano (Human Genome Project, HGP), avviato nel 1990 e dichiarato concluso nel 2003, ha rappresentato uno spartiacque. Da 20 anni a questa parte possediamo un “codice sorgente” delle 3 miliardi di coppie di basi nucleotidiche e dei circa 25,000 geni che costituiscono il genoma di Homo sapiens, a cui è possibile accostare i codici genetici di singoli individui per capire quali basi sono uguali e quali diverse. L’impatto di questa tecnologia in medicina, nell’ottica di una medicina di precisione, è enorme. Parliamo della possibilità di comprendere meglio le basi biologiche di centinaia di malattie rare, di caratterizzare lo spettro delle mutazioni somatiche dei tumori, e non da ultimo di stabilire punteggi di rischio per predire la prognosi di diverse patologie.
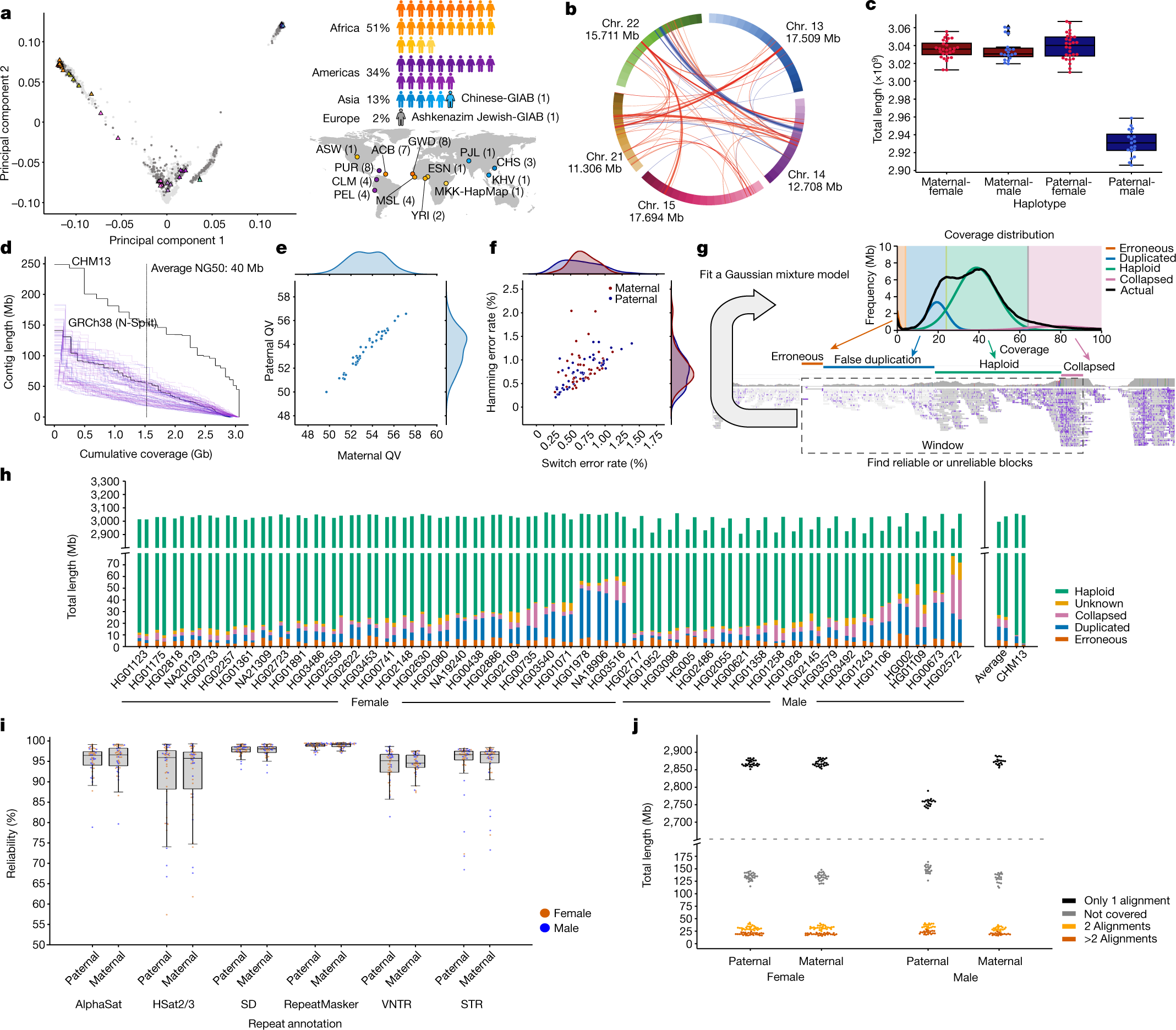
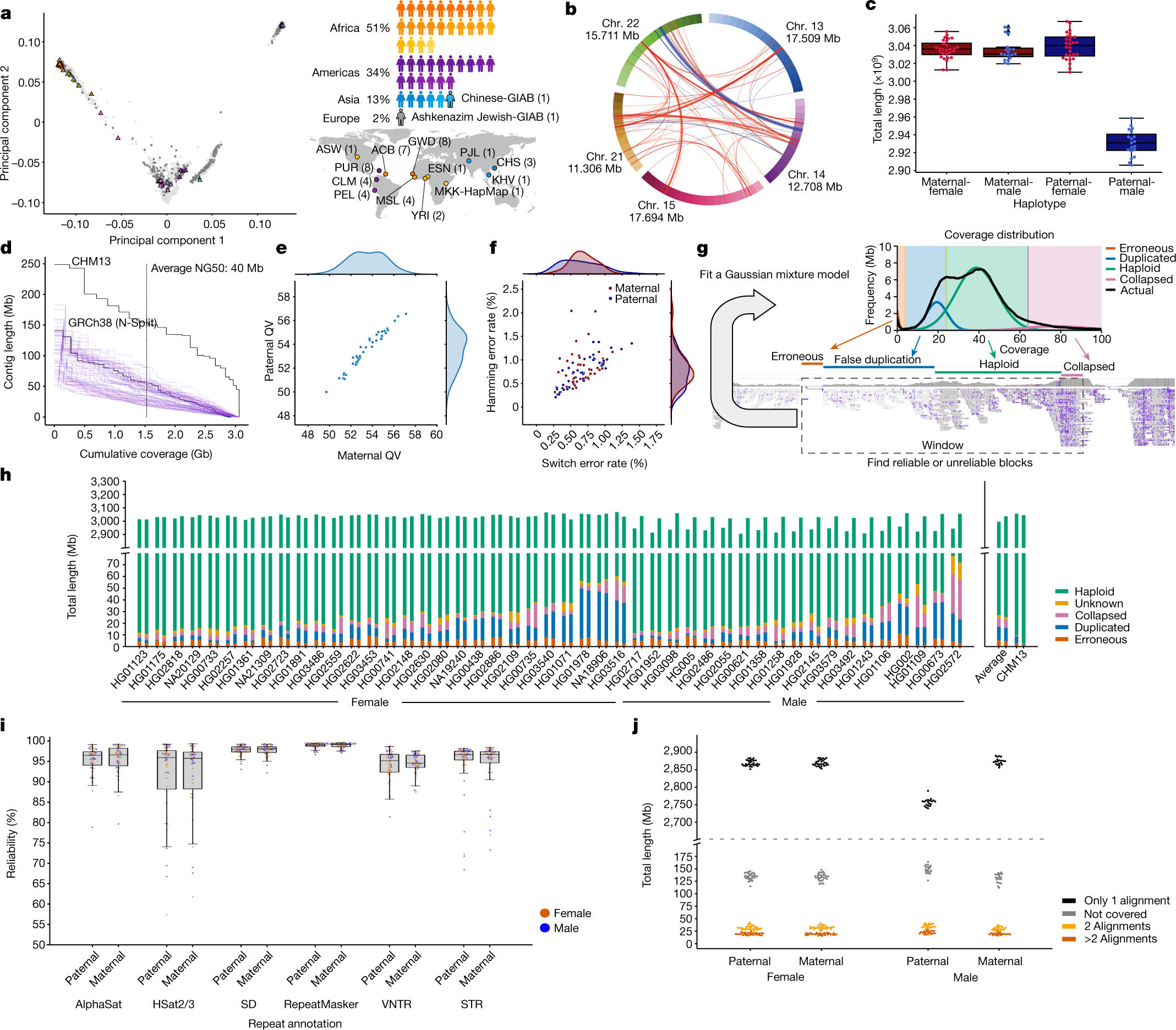
Oggi, con il manifesto dello Human Pangenoma, apparso sulla rivista Nature, si apre la possibilità di misurare il genoma umano di ognuno di noi in modo ancora diverso, per mappare ancora meglio la diversità genetica di Homo sapiens. Ne parliamo con Luca Pagani, professore associato di antropologia molecolare all’Università di Padova, dove si occupa di studiare la diversità genetica umana delle popolazioni.
Un breve ripasso da scuola superiore: in ogni cellula umana c’è un nucleo che contiene 23 coppie di cromosomi, i quali sono costituiti da una doppia elica di DNA attorcigliata su se stessa. Al suo interno, i nucleotidi, le basi, sono quattro – G=guanina, A=adenina, C=citosina, T=timina – e accoppiandosi e mettendosi in fila a coppie una accanto all’altra costituiscono appunto ila doppia elica. I geni invece sono coppie di basi che messe insieme svolgono una certa funzione nel nostro organismo e che se posseggono delle mutazioni patogeniche possono indurre una disfunzione, anche grave. “Chiaramente – precisa Pagani – in genetica utilizziamo la parola “mutazioni” con accezione neutrale. La maggior parte delle mutazioni che emergono confrontando il genoma di ognuno di noi con quello del 2003 sono non patogeniche, cioè non significano una modifica di un gene chiave che determina una disfunzione dell’organismo. È ovvio tuttavia che possedere tutta la lista di mutazioni non patogeniche può aiutarci a capire quali sono quelle fisiologiche e quelle che non lo sono.”
Ci rendiamo dunque facilmente conto che parliamo di stringhe di DNA lunghissime da “leggere”.
Nei 20 anni successivi al completamento dello Human Genome Project, sempre nuove tecnologie di sequenziamento (si parla di Next Generation Sequencing, NGS) sono riuscite ad abbattere di circa 200.000 volte i tempi e i costi delle analisi genomiche, che oggi sono alla portata di tutti. L’idea è questa: invece di sequenziare, cioè leggere, l’intera stringa di oltre 3 miliardi di lettere, si è capito che era sufficiente “tagliuzzare” il genoma in gruppi di 100 basi e accostarle come nel gioco del Memory accanto alla stringa di riferimento sequenziata nel 2003, per capire dove collocarli, ossia dove fosse situata quella stringa di basi ed individuare eventuali differenze. Se per ogni richiedente fosse necessario leggere tutti i 3 miliardi di lettere del suo genoma in un’unica sequenza, i tempi sarebbero lunghissimi, non gestibili, e prevederebbero conseguenti costi elevatissimi, fuori della portata di tutti.
Questo approccio rivoluzionario, sviluppato intorno al 2008, si basa sul fatto che esista per l’uomo il codice completo delle oltre 3 miliardi di basi, in un’unica stringa, che rimane il riferimento per comparare i gruppi di 100 basi provenienti dal genoma di ognuno di noi. Molte specie animali esotiche non sono mai state sequenziate, e pertanto richiedere un sequenziamento “ex novo” completo costa oggi molto di più rispetto a sequenziare il DNA umano. Parliamo di 300 euro contro un ordine di grandezza di 50 mila euro.
….segue


{kind=link}
{kind=link}